Im folgenden findet ihr hier meine Masterarbeit. Die Arbeiten dazu habe ich am Institut für Molekulare und Zelluläre Anatomie der Uniklinik RWTH Aachen unter Aufsicht von Rudolf Leube und Reinhard Windoffer durchgeführt. Ich stelle meine Arbeit hier hoch, in der Überzeugung, dass das an der Universität erlangte Wissen ein Gemeingut darstellt, welches der breiten Öffentlichkeit zur Verfügung gestellt werden muss. Forschung, wie sie an Universitäten durchgeführt wird, ist nur mit Hilfe öffentlicher Zuwendungen z.B. durch entsprechende Förderprogramme der Regierung möglich, welche steuerlich finanziert werden. Aus diesem Grund hat die Öffentlichkeit ein Recht auf den freien Zugang zu dem erlangten Wissen und dieses Wissen sollte ihm nicht durch das Closed Access Monopol akademischer Verlage vorenthalten werden können! Wissen, das mit Hilfe gesellschaftlicher Mittel gewonnen wurde, sollte nicht privatisiert werden dürfen, was heute in vielen Fällen faktisch der Fall ist. Durch das Hochladen dieser Arbeit möchte ich meinen Dank an all die Menschen zum Ausdruck bringen, welche mir diese wundervolle Chance ermöglicht haben, an der Forschung beteiligt zu sein. Danke!
I Abstract
Cells are highly sensitive to physical and mechanical cues arising within the microenvironment, which have important effects on cellular processes and subsequent biological responses. The cytoskeleton plays a pivotal role in sensing biomechanical cues and translating it into biochemical signals but it is also of vital importance as a stress-bearing structure for maintaining cell integrity and stability by reorganizing single cytoskeletal elements upon mechanical stimulation. The keratin intermediate filament network contributes to the mechanical resilience of epithelial cells by undergoing a continuous cycle of remodeling in response to mechanical forces. Alterations of the keratin dynamics are difficult to quantify in cells grown under classical culture conditions since the cell shape is transitory due to uncontrollable cell polarity, migration and division. Adhesive micropatterns enable control over cell shape and function. This work explores the possibility to manipulate cell shape and size through cell micropatterning techniques in order to enable an easier quantification of the keratin filament distribution.
In the course of the experiments, stencil patterning was established as the preferred method of choice due to its superior performance in fabricating accurately shaped micropatterns with a uniform and homogeneous protein coating. Micropatterns with different sizes and shapes were designed and the optimal sizes and shapes for a high yield of single cells of selected cell lines was ascertained. The results obtained from normalized cells indicate that the keratin intermediate filament network does not respond to local geometrical cues because differently shaped cells revealed similarities in the average distribution of the keratin intermediate filaments. Nonetheless, the results demonstrate the utility of cell micropatterning for quantitatively assessing the contribution of the cytoskeleton to cellular behavior. The establishment of stencil patterning and the subsequent results of this work motivate further extensive investigations of the keratin intermediate filament network, as well as its interplay with other cellular components.
[nextpage title=”1 Introduction”]
1 Introduction
In recent years, it has become increasingly evident that physical and mechanical cues generated intrinsically within the cell in response to its microenvironment, and extrinsic cues imposed upon the cell by the microenvironment are as critical as biochemical cues in regulating cellular structure and function (Fletcher and Mullins, 2010; Hersch et al., 2013; Janmey and McCulloch, 2007; Krieg et al., 2008; Provenzano and Keely, 2011; Théry et al., 2005).
Cells are able to sense their microenvironment and its variances through a vast arsenal of intracellular and extracellular receptors, which upon activation can induce different downstream signaling events including the activation of signaling networks with subsequent changes in gene expression, recruitment of biomolecules and reorganization of the cytoskeleton,. a scaffolding composed of three types of cross-linked filamentous networks together ensuring intracellular processes, structural integrity, and cell motility: actin microfilaments (MF), microtubules (MT), and intermediate filaments (IF).
1.1 Intermediate filaments
The human IF protein family is encoded by approximately 70 genes (Hesse et al., 2001), classified into six distinct types based on their similarities in gene structure or sequence homology and are expressed in a tissue type-dependent, cell type-dependent, differentiation program-dependent, and context-dependent fashion. IFs have been shown to play a crucial role in numerous cellular processes for maintaining cell and tissue integrity and have been implicated in playing a key role in stress response (Hyder et al., 2011; Kim and Coulombe, 2007; Pallari and Eriksson, 2006).
The most prominent types of IFs in epithelial cells are type I (acidic) keratins and type II (neutral-basic) keratins. Equal amounts of type I and type II polypeptides form heterodimers, which assemble into obligate heteropolymers. The keratin intermediate filaments (KIFs) form a cytoplasmatic network braiding the nucleus and extending out to the cell periphery. They are connected to the ECM through an anchorage to hemidesmosomes and to adjacent epithelial cells through desmosomes, thus ensuring the integrity and mechanical stability of both the single epithelial cells (Ramms et al., 2013) and of the epithelial tissue as it is evident in a number of keratin-associated disorders, where specific keratin mutations predispose cells to cellular stress and to epidermal fragility (Lane and McLean, 2004; Rugg and Leigh, 2004), resulting in various skin diseases such as Epidermolysis bullosa (Coulombe et al., 2009; Russel et al., 2004). Yet despite the importance of IFs for the maintenance of the biomechanical integrity of cells and tissue, their contribution to intracellular mechanics mostly remains unknown. Studies exploring the mechanical properties of IFs have shown that single IFs are quite soft, strikingly extensible, and withstand greater mechanical deformation than either microtubules or actin filaments (Fudge et al., 2003; Fudge et al., 2008; Janmey et al., 1991; Kreplak and Fudge, 2007). Cells in epithelia are exposed to tremendous mechanical stresses, for example frictional forces or strain, and as the predominant cytoskeleton in epithelial cells KIFs prove to be of great importance for the biomechanical integrity and stability of epithelial cells in response to mechanical resilience. Epidermal keratinocytes lacking type I and type II keratins, respectively, are significantly softer and less viscous than WT cell lines (Ramms et al., 2013).
The contribution of both actin and microtubules to mechanical resilience of cells and their polarity, the dynamics of their assembly and the associated motors and regulatory proteins have been investigated in great detail (Blanchoin et al., 2014, Fletcher and Mullins, 2010). Both actin and MT networks are composed only of one highly conserved globular protein, actin and tubulin, respectively. The basis for the remodeling of both networks is the asymmetry of the subunits and their dependency on chemical energy derived from hydrolysis. In contrast, the molecular mechanisms governing the polymerization of IFs and their dynamics are not well understood but its uncovering is of vital importance to understand cellular function and various human genetic disorders. IF self-assembly does not require chemical energy and due to the symmetric composition of their tetrameric subunits IFs lack polarity. High-resolution time-lapse imaging of cells transfected with fluorescently tagged keratin revealed a high degree of keratin network dynamics in cultured cells, enabling the establishment of a reference model for the KIF assembly and disassembly, termed as the keratin cycle (Windoffer et al., 2011). The keratin cycle (Fig. 1) is subjected to a protein biosynthesis-independent turnover and begins with the nucleation of soluble keratin particles, referred to as KIF precursors, in the cell periphery in close vicinity to focal adhesions followed by elongation, subsequent integration into pre-existing peripheral KIF networks and bundling, revealing an inward-directed movement, which is proposed to rely mainly on actin microfilaments but also on microtubules. Some KIFs disassemble into soluble subunits that diffuse throughout the cytoplasm and can be re-utilized for another round of KIF assembly. Other KIFs exit the keratin cycle of assembly and disassembly and mature into a stable network braiding the nucleus and are anchored to hemidesmosomes and desmosomes, resulting in an increased mechanical resilience of cells (Strnad et al., 2002).
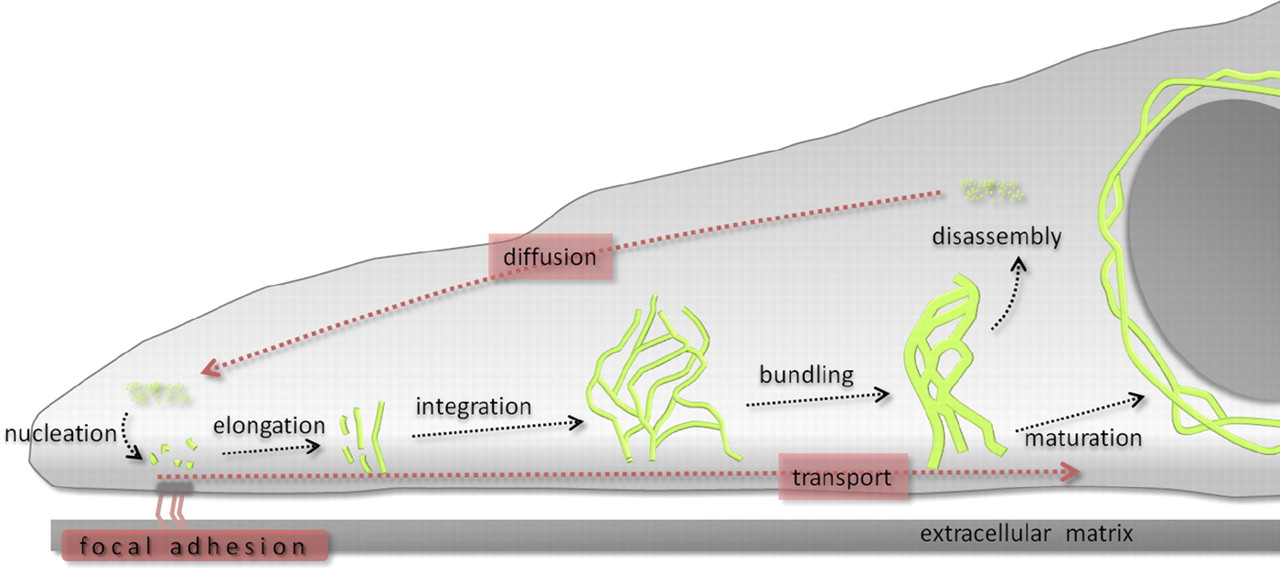
Recent advances towards image analysis methods have been made to determine the speed and direction of KIF motion and to identify locations of KIF polymerization and depolymerization at subcellular resolution (Moch et al., 2013). Put together, the keratin cycle of assembly and disassembly and the keratin motion analysis yield a strong model, which provides a framework to identify regulatory proteins altering the cycle and to investigate keratin modifications and the influences of mechanical forces on the keratin cycle and the cell.
In order to measure keratin dynamics and its variances after alternations of experimental setups attempts have been made for conventional 2D cell culture systems to improve the comparability of single cells by converting recorded single cell images into idealized round cells, thus subtracting out variances in cell morphology (Moch et al., 2013). Unfortunately, such sophisticated cell shape normalization techniques are prone to errors that may occur in normalizing migrating or contracting cells, are only suitable for round cells and labor extensive. Several methods have been developed to overcome these obstacles, cell micropatterning being one of the most promising developments enabling the manipulation of cell architecture, cell-extracellular matrix (ECM) and cell-cell interactions.
[nextpage title=”1.2 Cell micropatterning: a tool to regulate cell shape”]
1.2 Cell micropatterning: a tool to regulate cell shape
Cell micropatterning refers to a technique within cell culture methods whereby the assembly of ECM-proteins e.g. fibronectin (FN) on a specific surface is controlled in order to achieve an accurate positioning of cells and control over cell size and spatial arrangement. This requires the ability to fabricate adhesive regions which mediate cell attachment and non-adhesive regions preventing cell attachment. Adhesive regions, commonly called micropatterns, can be fabricated in different shapes and sizes and can have a resolution reaching micro-scale.
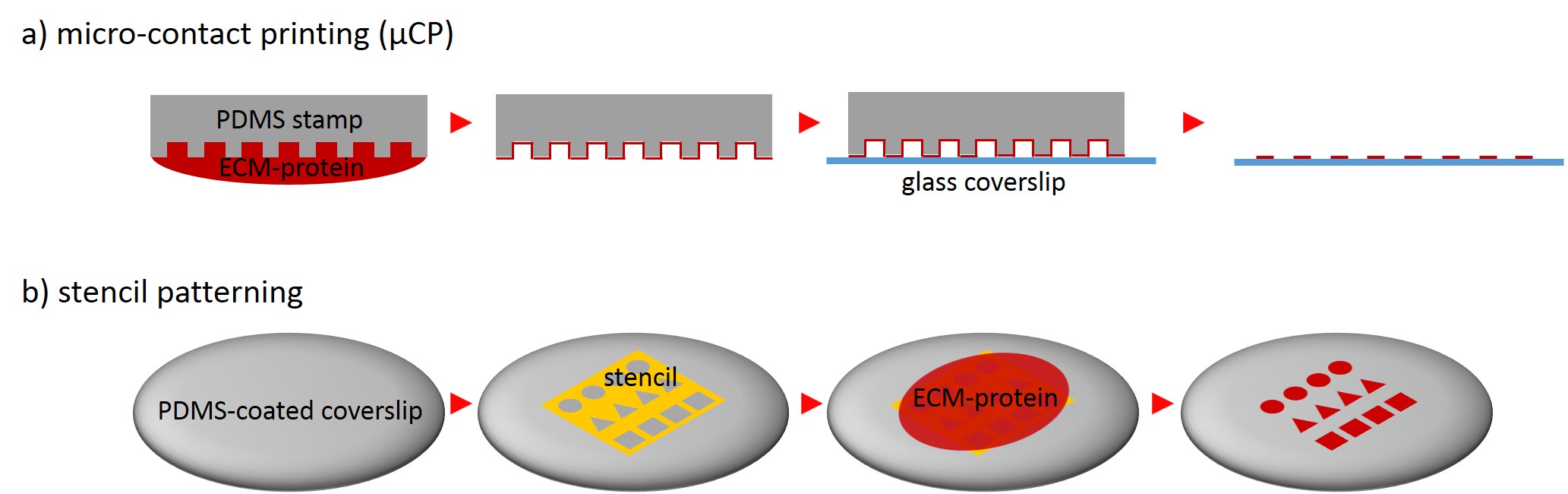
One of the techniques most used in order to fabricate micropatterns on a planar substrate is micro-contact printing (µCP), in which a polydimethylsiloxane (PDMS) stamp is fabricated based on a replica molding process by casting the liquid prepolymer of PDMS over a rigid master with patterned structures. After curing, the PDMS stamp is peeled off the mold, inked with a protein of choice and transferred onto the surface of a planar substrate by placing the microstructured, inked surface of the stamp in contact with the substrate. Upon removing the stamp, the desired pattern is left behind on the surface (Fig. 2 a). Alternatively, a stencil containing through holes can be used to fabricate micropatterns. The stencil can be fabricated either by casting a thin film of the liquid material to be used directly on the master without covering it wholly so that through holes can form or by depositing a curable material on the edges of a PDMS stamp placed on a planar substrate. Via capillary suction the gaps between the stamp and the substrate is filled. After curing and the removal of the stamp, the stencil can be peeled off and applied to the final substrate to be used. Adhesive proteins deposited on top of the stencil can come in contact with the underlying substrate only at regions left exposed by the though holes, resulting in the formation of adhesive micropatterns (Fig. 2 b).
The protein of choice can range from adhesive ECM proteins, e.g. fibronectin, collagen or laminin to amine-terminated self-assembled monolayers (SAMs) or Arg-Gly-Asp (RGD) peptide.
Upon seeding cells on a micropatterned substrate cells adapt a similar shape and size as the adhesive micropatterns thus enabling the manipulation of cell architecture, cell-ECM and cell-cell interaction.
In the past decades it has been revealed that cell morphology regulate different cellular processes like differentiation and cell fate, cell cycle control, proliferation, apoptosis and migration (Chen et al., 1997; de Juan-Pardo et al., 2006; Huang et al., 1998; Huang and Ingber, 2000; Ingber et al., 1995; LeDuc and Bellin, 2006, Parker et al., 2002). For example, Chen et al. (1997) demonstrated that human and bovine capillary endothelial cells were limited from spreading when micropatterns decreased in size and that cell fate shifted from growth to apoptosis, regardless of the type of adhesive protein used to promote attachment. Recent studies demonstrate how far-reaching geometric influences can be: It has been hypothesized that changes in cell architecture and associated changes in nuclear shape cause changes in chromatin structure and organization and thus alternating transcriptional activity. Stiles et al. (2013) aimed to uncover the influence of different cell shapes on global gene expression patterns of human coronary artery endothelial cells. Stiles and colleagues shown that morphological restriction in general significantly affect global gene expression patterns compared to non-restricted cells. These and many other examples demonstrate the influence of the extracellular physical environment in regard to cell response. Apart from their usefulness in exploring alternations in cellular morphology and physiology adhesive micropatterns ensure reproducibility, enable the detection of drug effects and facilitate automated image acquisition and analysis.
[nextpage title=”1.3 Objective”]
1.3 Objective
The objective of this work was to establish a cell micropatterning method that warrants reproducible cell shape, enabling a fast and accurate quantification of cytoskeletal organization. In order to achieve this goal, following major issues were addressed:
-Micropatterns with different shapes and sizes were designed and fabricated.
-µCP and stencil patterning were tested and compared in regard to their performance in fabricating micropatterns and in printing a uniform protein coating.
-The behavior of two selected cell lines was observed on micropatterned substrates.
-The optimal micropattern size and shape for a high yield of single cells was detected.
-The KIF distribution was assessed in normalized cells.
-Heat maps were generated for normalized cells.
[nextpage title=”2 Materials and Methods”]
2 Materials and Methods
Given the complexity and the extremely wide scope of this part, I decided to leave out the description of the materials. Whoever would like to know the exact details, don’t hesitate to contact me! I will forward you the details with pleasure. Please write a message by means of the contact form on my website.
2.2 Methods
2.2.1 Lithographic techniques
2.2.1.1 Fabrication of photolithography mask
The following method was done in cooperation with the Institute of Complex Systems 7: Biomechanics at the Jülich Research Centre. Different micropatterns for single cell analysis and bowtie-shaped micropatterns for cell-cell contact analysis were designed using the CleWin software.
The micropatterns were written on a blank photomask coated with a chromium layer and the negative photoresist layer SAL 601 by using the electron-beam lithograph EBPG-5H. The post-exposure bake of photomask was performed for 2 minutes at 105°C. Uncross-linked photoresist was removed using a 1 to 2 dilution of AZ 400K in DI water. Photomask was hard-baked for 15 min at 100°C, 15 min at 130°C and 15 min at 150°C successively. To remove residual layers of photoresist areas photomask was descummed for 10 to 120 s. Wet etching followed for 1 min in a chrome etching solution with a subsequent wash step in DI water. Finally, photomask was dried under a stream of nitrogen gas and exposed to oxygen plasma.
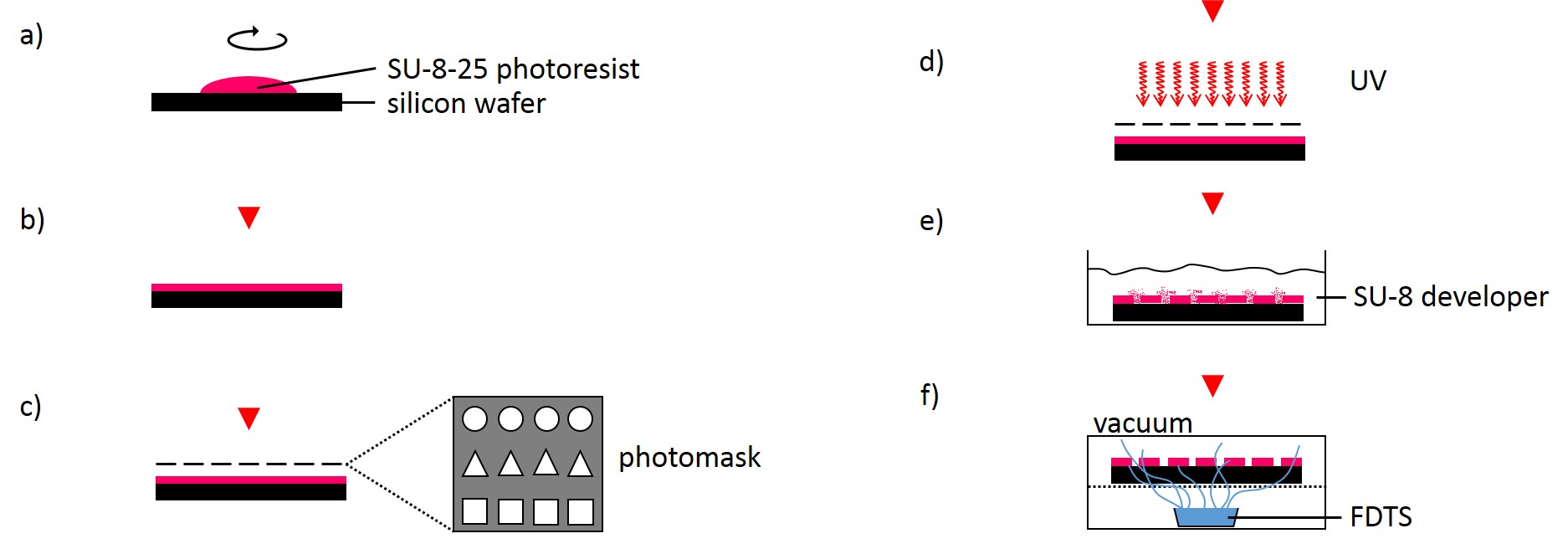
2.2.1.2 Fabrication of microstructured silicon master
The following method was performed by the Institute of Complex Systems 7: Biomechanics at the Jülich Research Centre. A silicon wafer was preheated at 180°C for 30 min and overlayed with a 25 µm thick layer of SU-8-25 photoresist (Fig. 3). Subsequently, the photoresist layer was soft-baked for 2 min at 65°C followed by slow ramp heating to 90°C within 5 min and additional baking of the photoresist at 90°C for 2 min. Using ultraviolet-photolithography at 346 nm with a source power of 7 mW for 25 s, the structures of the photolithography mask were transferred into the photoresist layer. The wafer was post-baked for 1 min at 65°C followed by slow ramp heating to 90°C within 5 min and additional baking for 1 min at 90°C. Uncross-linked photoresist was removed using SU-8 developer for 6 min. Finally, the photoresist was hard-baked for 30 min at 180°C. The SU-8-25 wafer was then silanized by exposure to vapor of FDTS in vacuum for 30 min at room temperature in order to prevent it from adhering to the PDMS during stamp fabrication.
2.2.2 Micropatterned substrate fabrication
2.2.2.1 Micro-contact printing
The first step for the fabrication of microstructured substrates using the µCP method consists of the fabrication of a microstructured master (see 2.2.1.2). The silicon master was cleaned under a stream of nitrogen. Sylgard® 184 Silicone Elastomer Base and Sylgard® 184 Silicone Elastomer Curing agent were mixed in a 10:1 ratio by thoroughly stirring. After removing air bubbles by degassing the solution in a vacuum desiccator the elastomer was poured over the master and cured at 60°C for 2 h (Fig. 4). The elastomer was peeled off the silicon master and small stamps were excised from the regions of interest. A drop of 50 µg/ml FN in PBS or 45µg/ml Collagen I in acetic acid mixed with 0.3% of an antibody of choice in PBS and acetic acid respectively was placed onto the microstructured surface of the PDMS-stamp. The ink was incubated for 1 h at RT. Subsequently, the ink drop was aspirated and the inked surface dried under a stream of nitrogen gas for 30 s. The PDMS-stamp was inverted and placed onto a gamma irradiated glass coverslip of a glass bottom dish immediately for 3 s and 30 min respectively. A 2 g weight was placed onto the PDMS-stamp in order to promote good contact with substrate and to stabilize the construct. The construct was carefully disassembled. Non-printed areas were blocked with a 1% Pluronic® F-127 in ddH2O for 1 h at 4°C in order to prevent cell adhesion. Subsequently, the substrate was carefully rinsed tree times with PBS.
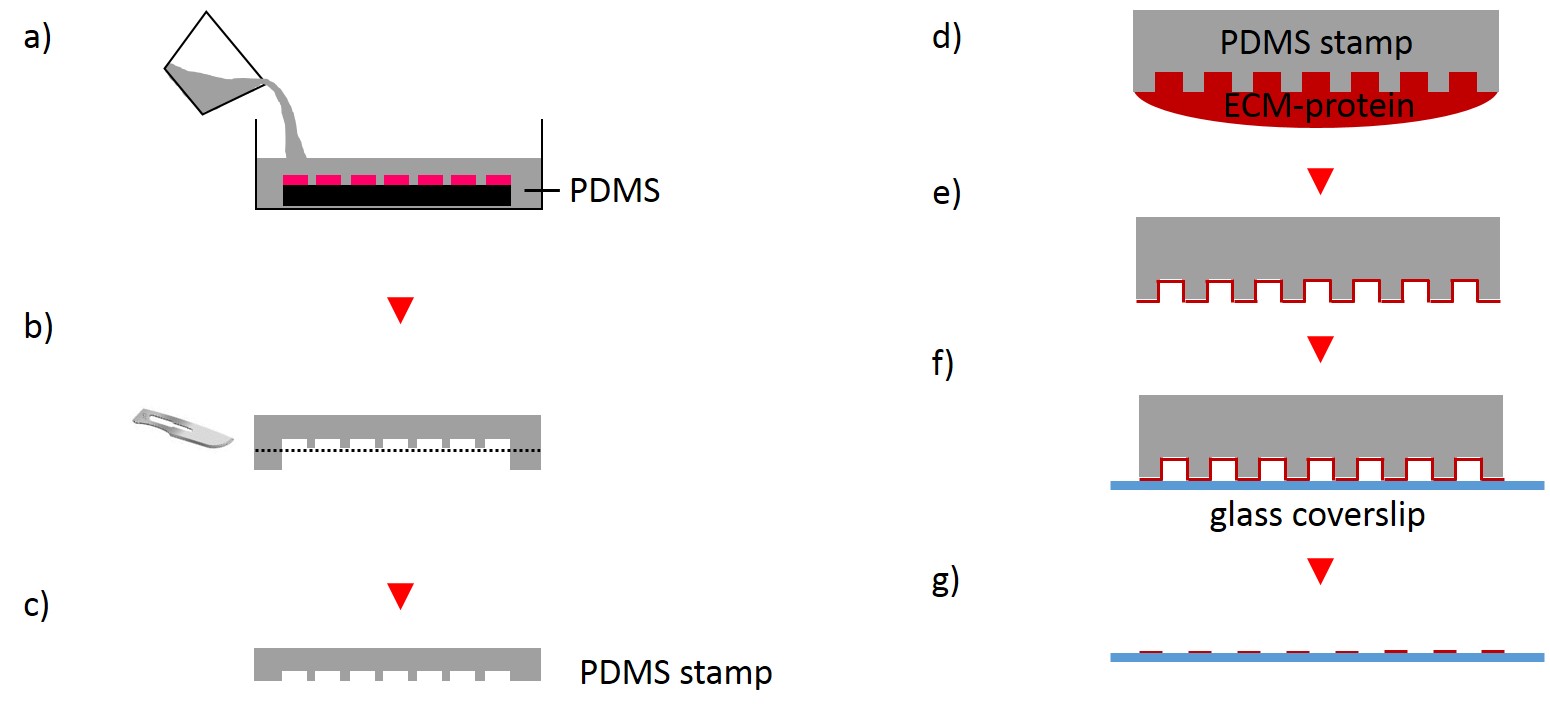
2.2.2.2 Stencil patterning
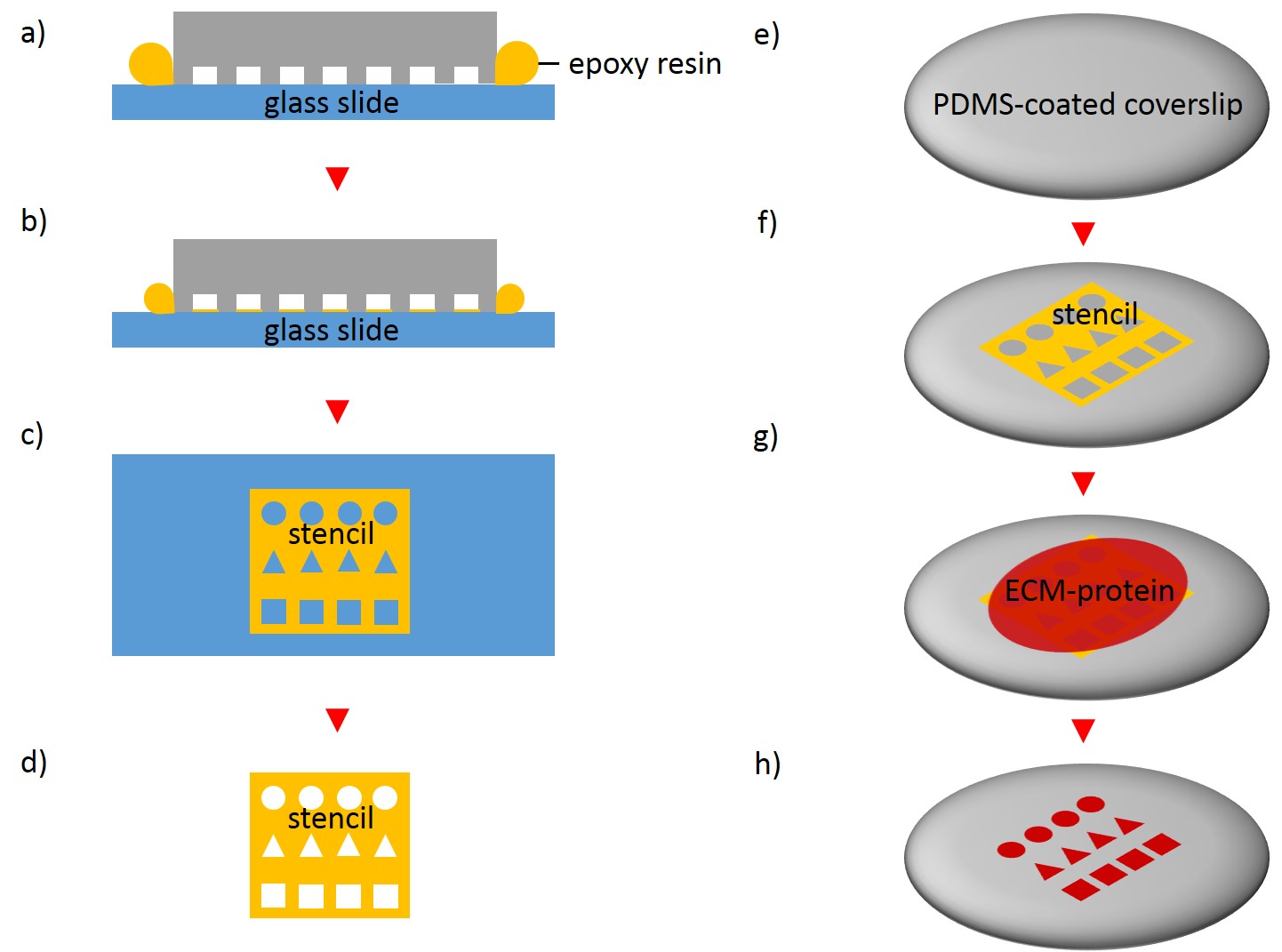
In order to fabricate microstructured substrates through the usage of stencils PDMS-stamps were fabricated as described above (see 2.2.2.1). In the first step, coverslips were spin coated with a thin layer of PDMS (base to curing agent 10:1) for 7 s at 2500 rpm. Coated coverslips were mounted on the outer side of culture dishes with holes of approx. 20 mm diameter in a leak proof manner, the coated side facing the interior part of culture dishes. PDMS was cross-linked for 16 h at 60°C. Alternatively, cross-linked PDMS-coated coverslips were places in microwell plates. In the second step, PDMS-stamps were placed onto a sellotape adhered to a glass slide. Uncured epoxy resin was placed at the stamp boundaries and dispensed by capillary suction between stamp and sellotape (Fig. 5). Curing was performed through exposure to UV on a transilluminator table for 40 min. Stamps were removed and the microstructured stencils were peeled off the sellotape. Next, the stencil was placed on a PDMS-coated coverslip. A drop of 50 µg/ml FN in PBS or 45 µg/ml Collagen I in acetic acid mixed with 0.3% of an antibody was placed in top for 60 min. Trapped air bubbles were removed by degassing the construct for 2min. Afterwards, the solution was discarded and the membrane peeled off carefully from the elastomer substrate. Non-printed areas were blocked with 1% Pluronic® F-127 in ddH2O for 10 min at 37°C in order to prevent cell adhesion. Finally, the substrate was carefully rinsed tree times with ddH2O.
2.2.2.3 CYTOOchips™
A Starter’s CYTOOchip bears four different fluorescently labeled FN micropatterns (disc, crossbow, H and Y) in three sizes (1600 µm², 1100 µm², 700 µm²). The CYTOOchips were placed in independent wells of a 6 well plate. It was made sure that the CYTOOchips were placed correctly so that the CYTOO logo could be read the right way in the lower right of the chips. Since CYTOOchips were already patterned and coated by the manufacturer cells were ready to be dispensed into each well containing a CYTOOchip.
2.2.3 Cell culture
2.2.3.1 Cultivation and passaging adherent cells
Human vulva carcinoma-derived A431 subclone AK13-1 cells, producing human keratin 13-EGFP, and immortalized human keratinocyte-derived subclone HaCaT B10 cells, producing human keratin 5-EYFP cells, were grown in DMEM supplemented with 10% FCS Gold at 37°C and 5% CO2. In addition, 500 µg/ml G418 was added to the culture medium of HaCaT B10 cells. For subculturing purposes, cells were passaged after reaching 70-90% confluence, taking approximately up to 2 to 3 d. Medium was aspirated and cells were washed shortly with DPBS. Cells were trypsinized by adding a trypsin/EDTA solution and incubating them for about 15 min at 37°C and 5% CO2. Fresh medium was added and trypsinized cells were pipetted up and down several times to ensure a uniform cell suspension. To ensure a uniform, single-cell suspensions cell strainers with a pore size of about 40 µm were used. Cells were pelleted by 1000 rpm for 3 min, the supernatant was aspirated and cells were resuspended in fresh media, and transferred into a fresh flask containing fresh medium, ensuring an appropriate dilution concentration.
2.2.3.3 Cryopreservation of mammalian cells
For freezing procedures, medium was aspirated and confluent grown cells washed shortly with DPBS and trypsinized with trypsin/EDTA at 37°C and 5% CO2 until cells detached. Fresh DMEM medium supplemented with 10% FCS Gold was added and cells were pipetted up and down several times to ensure a uniform cell suspension. Next, cells were pelleted by low speed centrifuging at 1000 rpm for 3 min. Medium was discarded and cells were resuspended in pre-cooled freezing medium and aliquoted into cyrotubes. The cyrotubes were incubated in a vessel containing isopropanol at -80°C overnight and transferred into liquid nitrogen for long-term storage.
For thawing cells, cyrotubes were swayed in a 37°C pre-warmed water bath for rapid thawing. Subsequently, cells were resuspended in pre-warmed fresh DMEM medium supplemented with 10% FCS Gold and seeded into a new flask.
2.2.3.4 Cell seeding on micropatterns
After the fabrication of micropatterned substrates cells were collected by trypsinization as described above. Cells were diluted to a concentration of 15000 cells/ml or less in fresh DMEM medium supplemented with 10% FCS Gold, dispensed onto micropatterned substrates and incubated at 37°C and 5% CO2. Depending on the cell line, after about 2-7 h non-adherent cells were removed by gently aspirating the medium without letting the coverslips dry out entirely. DPBS was added and aspirated off. It was checked under the microscope for floating cells. If a large number of floating cells were observed, washing procedure was repeated. DPBS was replaced with fresh medium. Thereafter, it was checked repeatedly for full cell spreading.
2.2.4 Immunofluorescence staining
After cell seeding and spreading on micropatterns, medium was aspirated and substrates washed briefly in pre-warmed PBS. Cells were fixed for 5 min at -20°C in methanol and subsequently for 20 s at -20°C in acetone. After fixation, substrates were air-dried until acetone evaporated and then rinsed with PBS for 10 min at RT. Immunostaining was performed at RT for 1 h in primary antibody. Next, cells were rinsed tree times with PBS and then incubated for 1 h at RT with the secondary antibody in the dark. After three times washing with with PBS for 10 min cells were rinsed in ddH2O and finally mounted with Mowiol supplemented with 1 µg/ml DAPI on microscope slides.
2.2.5 Image acquisition and analysis
Immunofluorescence staining was assessed by an Axio Imager M.2 microscope equipped with an ApoTome.2 unit using an Axiocam MRm camera and with EC Plan-Neofluar 10x/0.30 Ph 1, Plan-Apochromat 20x/0.8, EC Plan-Neofluar 40x/0.75 and Plan-Apochromat 63x/1.40 Oil DIC M27 objectives. In general, best automatic settings were chosen for exposure.
Fluorescence recording was also done by confocal laser scanning microscopy using a LSM 710 Duo. When more than one fluorophore was used in a single sample, the emission spectra were adjusted to prevent significant overlap. Except for the comparison of fluorescence intensity, best automatic settings were chosen for laser intensity and gain. The diameter of the pinhole amounted to 1 AU.
The evaluation of pictures was done using the Zeiss ZEN 2009 software and the AxioVision software. Image processing was performed using the ImageJ-based image processing program FIJI. For generating heat maps CYTOO tooL for Image Processing-Reference cell, an ImageJ macro provided by CYTOO Inc., was tested.
[nextpage title=”3.1 Experimental results from lithographical techniques”]
3 Results
3.1 Experimental results from lithographical techniques
In order to regulate cell shape and subsequently the cytoskeleton, adhesive islands consisting of ECM-proteins and of different geometric shapes were generated. For this approach, geometric shapes of different sizes were designed and written on a photomask during a photolithography process and subsequently transferred onto a silicon master (Fig. 6 A). The silicon master was used for casting PDMS stamps, which in turn were used for substrate patterning either by µCP (Fig. 6 B) or by stencil patterning (Fig. 6 B) through the fabrication of stencils.

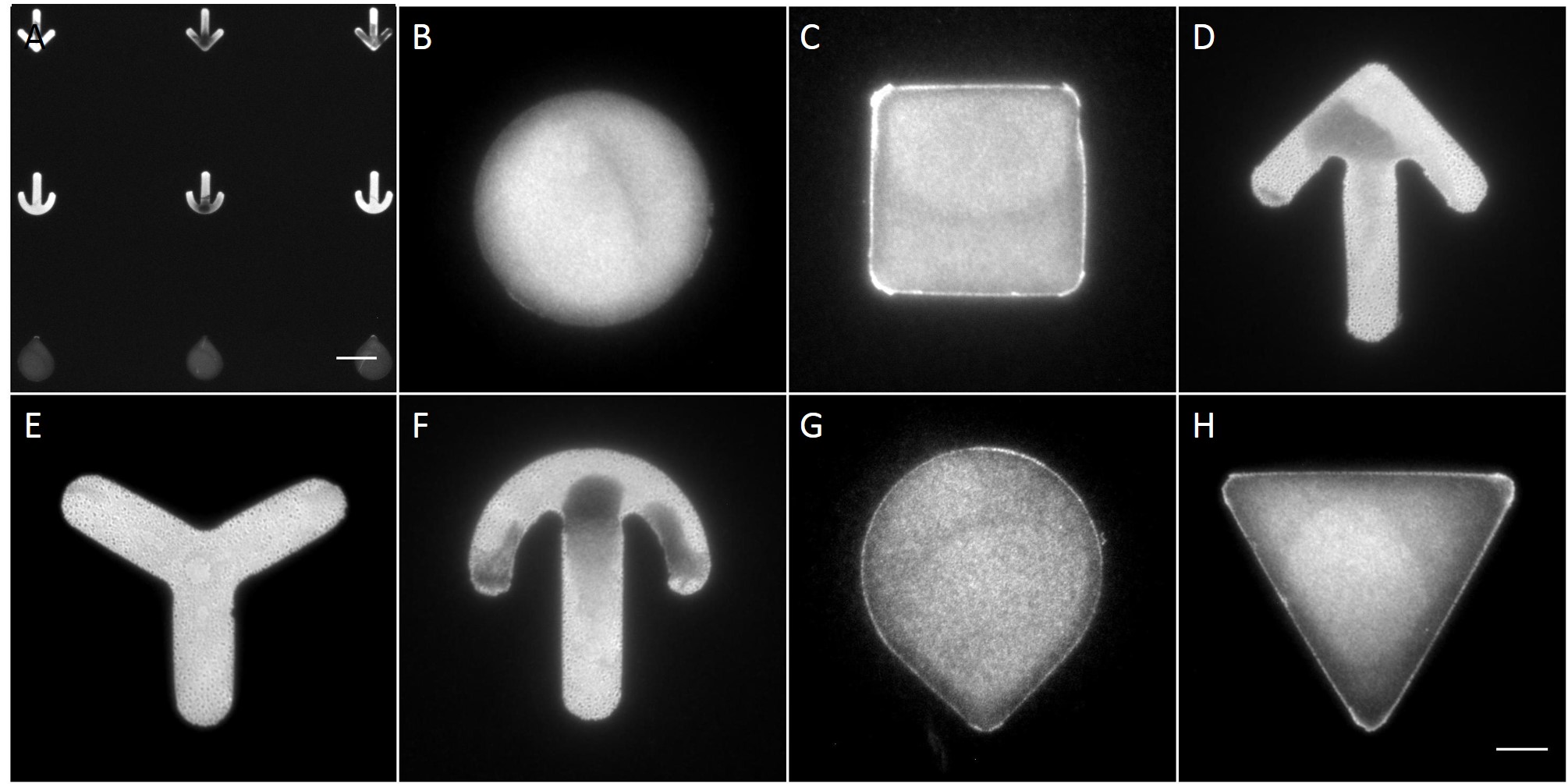
The microstructures are shown in Figure 7. By way of illustration, the micropatterns are shown as they are observed on stencils. Two types of micropatterns were designed: bowtie (Fig. 7 A) and single (Fig. 7 B) patterns. Each pattern was designed in four different sizes allowing an average cell spreading area of 1600 µm², 1100 µm², 900 µm² and 700 µm². The distance between the patterns amounted to approx. 150 µm in order to prevent cells from bridging the gap between the patterns.
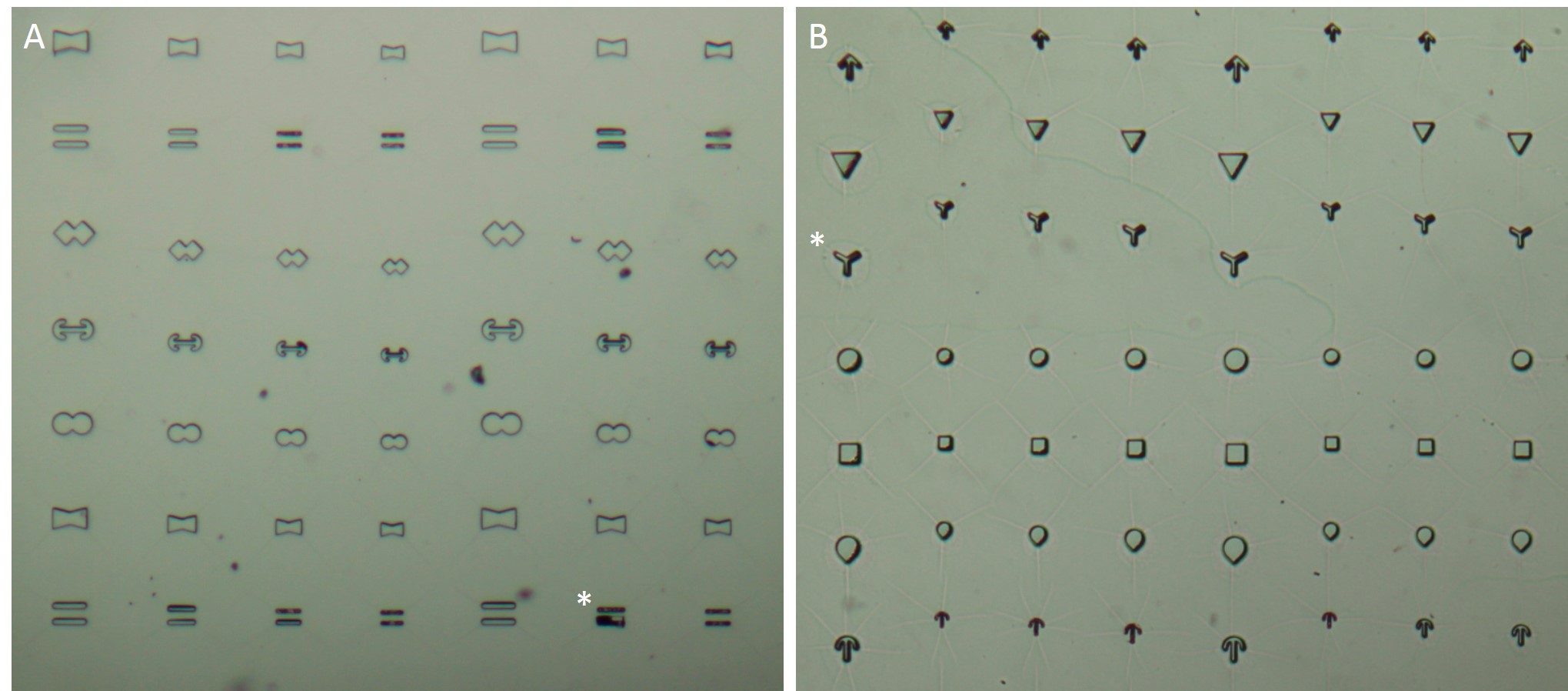
Most patterns were well molded and the holes spanned the entire stencil. However, some patterns (Fig. 7 *), especially those composed of thin lines, showed an insufficient or no through holes, which cause malformed or no micropatterns during stencil patterning.
[nextpage title=”3.2 Comparing µCP and stencil patterning”]
3.2 Comparing µCP and stencil patterning
In the scope of this work, unmodified glass slides were used for µCP. Stamp was incubated with a FN/fluorescently labelled secondary antibody ink solution for 1 h. After aspirating the solution and drying the ink, the microstructured stamp surface was placed in contact with the non-treated glass substrate for 1 min, 30 min and 1 h respectively. The resulting patterns are illustrated in Figure 8. Local inhomogeneity of protein density and malformed patterns was observed frequently. In some cases, a faint imprint indicated a small displacement of the stamp during the process. Other regions were covered completely with the ink solution. Overall, µCP with unmodified glass surfaces show an insufficient protein patterning.
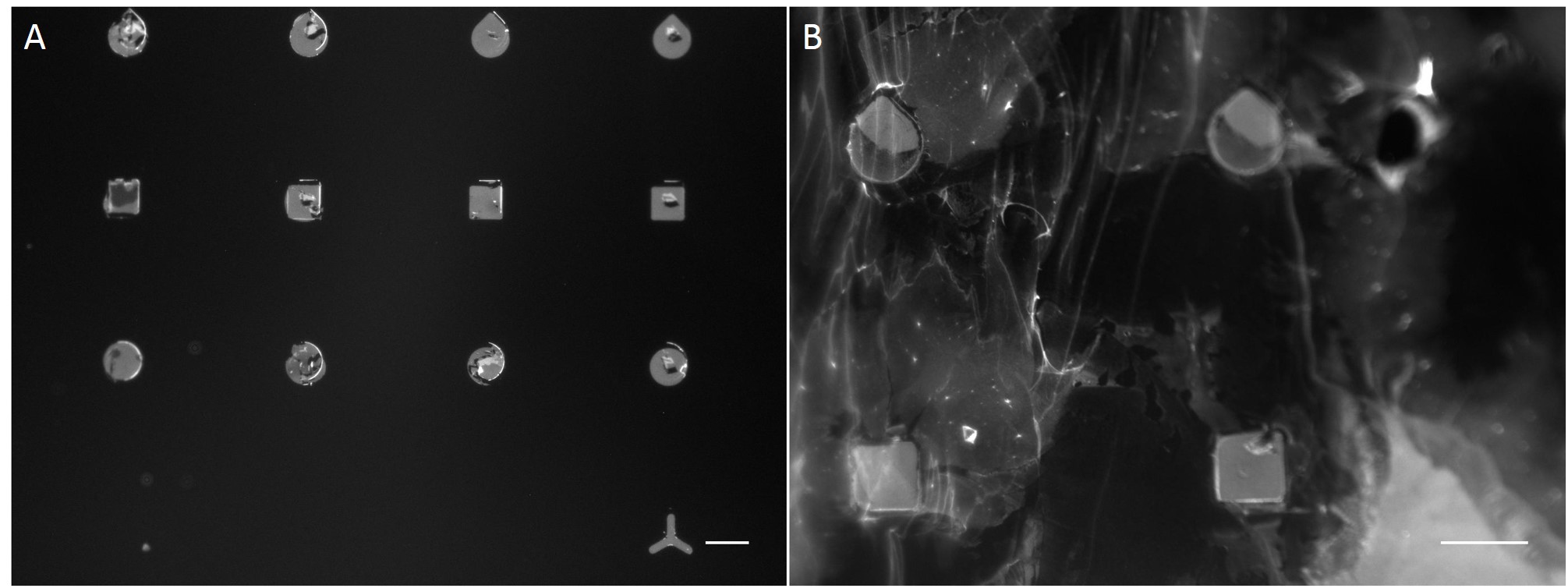
In order to achieve better results, a second printing method was tested. Stencil patterning is based on the usage of thin sheets comprising through holes of the desired pattern and size. The stencil is deposited on a silicone elastomer substrate and a coating solution is placed on top of the microstructured stencil. Areas outside the through holes are protected from coming into contact with the coating solution whereas regions with through holes are exposed to the solution and are locally modify.
Compared with the results of µCP this approach shows better performance in homogeneity of protein density and in fabricating accurate features (Fig. 9).
[nextpage title=”3.3 Behavior of cells on micropatterns”]
3.3 Behavior of cells on micropatterns
Due to the superior performance in protein patterning, stencil patterning was chosen for all subsequent experiments. After protein patterning, a subsequent incubation with the antifouling agent Pluronic® F-127 was performed in order to passivate non-printed regions and to allow specific adhesion of cells to the micropatterns. AK13-1 and HaCaT B10 cells were seeded at a density of 104 cells per cm2 and were maintained at 37°C and 5% CO2. Because cell attachment and full cell spreading time vary between the different cell lines, an adequate incubation time needed to be found accordingly. Generally, cells were checked in 2-4 h steps up to 24 h for cell attachment and spreading.
Both cell types on printed silicone elastomer substrates differed in shape and spreading behavior from cells on other substrates. While most cells attached rapidly on silicone elastomer substrates coated wholly with FN, on FN coated glass coverslips and on non-coated glass coverslips and reached about 50 µm in diameter, cells on micropatterned silicone elastomer substrates needed >6 h for cell attachment.
In order to minimize cell attachment time the passivation step with the antifouling agent Pluronic® F-127 was excluded since only a negligible number of attached cells were observed on non-coated silicone elastomer substrates and because Pluronic® F-127 is suspected to attach to micropatterns and thus prevent proper cell attachment and spreading. Cell micropatterning without the usage of Pluronic® F-127 resulted in a faster cell attachment (The time needed for cells to spread on micropatterns was approx. 24 h and remained unchanged despite the usage of conditioned cell culture media. To find out whether cells favored printed glass substrates over printed silicon elastomer substrates cells were tested for spreading time on CYTOOchips™ and on printed elastomer silicon substrates. A comparison revealed no notable changes in cell spreading time.
[nextpage title=”3.4 Normalized cell shape”]
3.4 Normalized cell shape
To investigate the role of geometric features on the KIF network and to identify the appropriate size of patterned features, AK13-1 cells and HaCaT B10 cells spread on micropatterns with an average cell spreading area of 700 µm², 900 µm², 1100 µm² and 1600 µm² were analyzed. After cell attachment and spreading an increased number of malformed micropatterns could be observed (Fig. 10). These were mostly micropatterns occupied by one or multiple cells, suggesting that the printed proteins were peeled off by the cells, resulting in pattern degradation. A retreat of the micropattern boundaries is observed, which correlates with the cell boundaries (Fig. 10 A’ and B’). As the overall shape of the patterned cells was unchanged, these cases were also documented and the effects of pattern degradation on the distribution of KIFs assessed as negligible or insignificant.
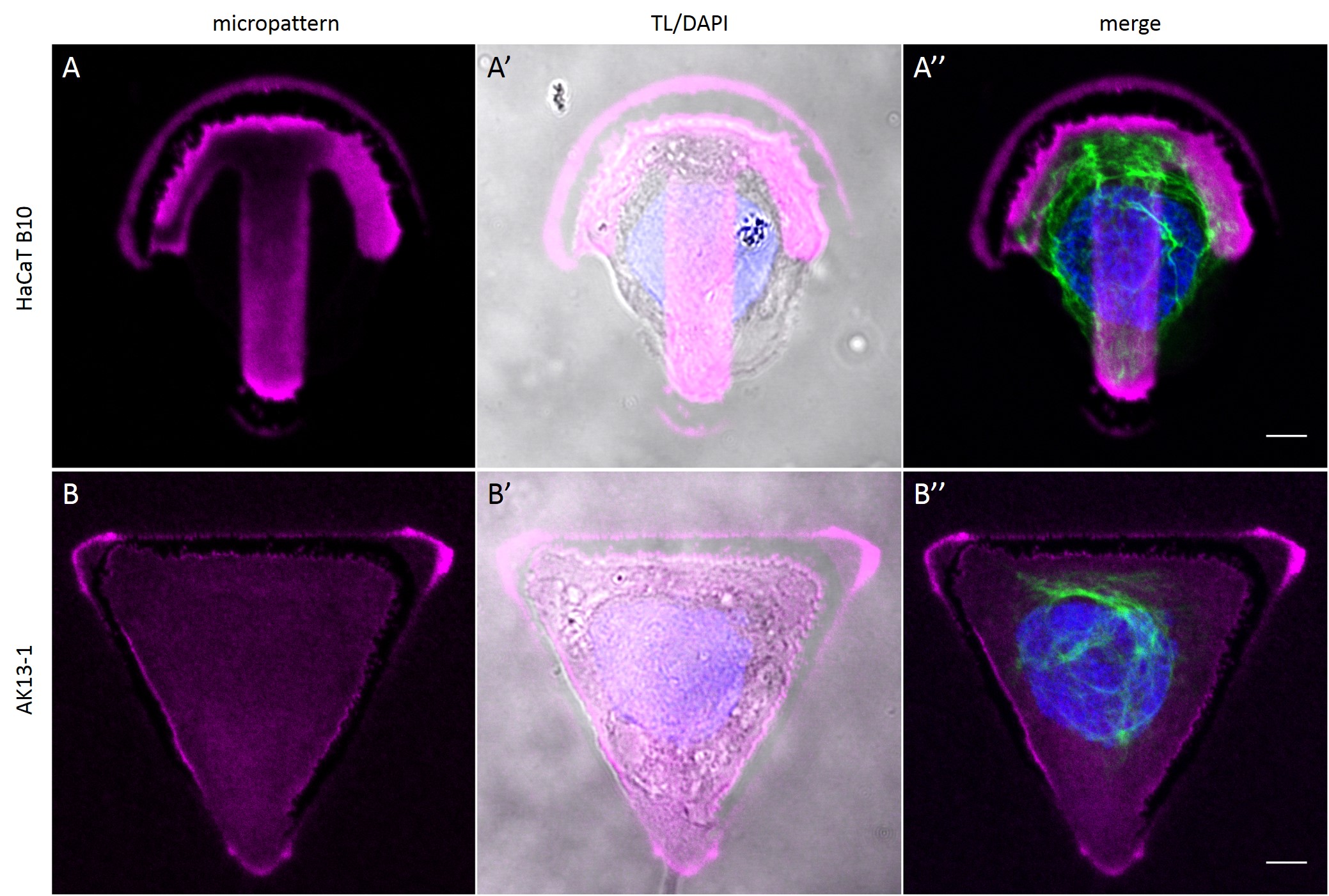
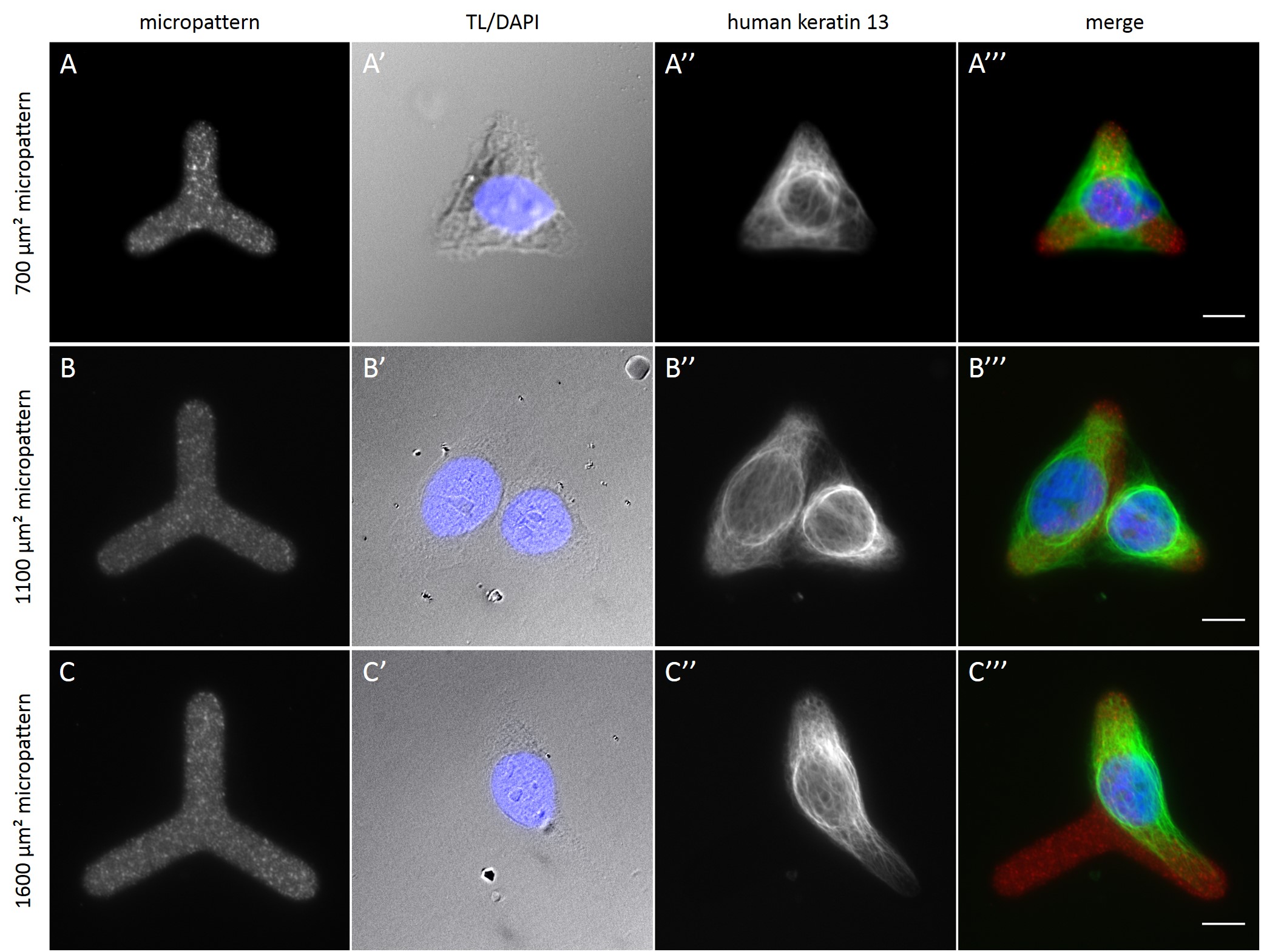
Fully spread single cells were found often on small micropatterns (Fig. 11 A-A”’) whereas an increase in micropattern size led to an increase in the number of cells per micropattern (Fig. 11 B-B”’) or to incomplete cell spreading of single cells (Fig. 11 C-C”’). Hence, less or no fully spread single cells could be found for many large micropatterns.
[nextpage title=”3.5 Distribution of KIFs in normalized cells”]
3.5 Distribution of KIFs in normalized cells
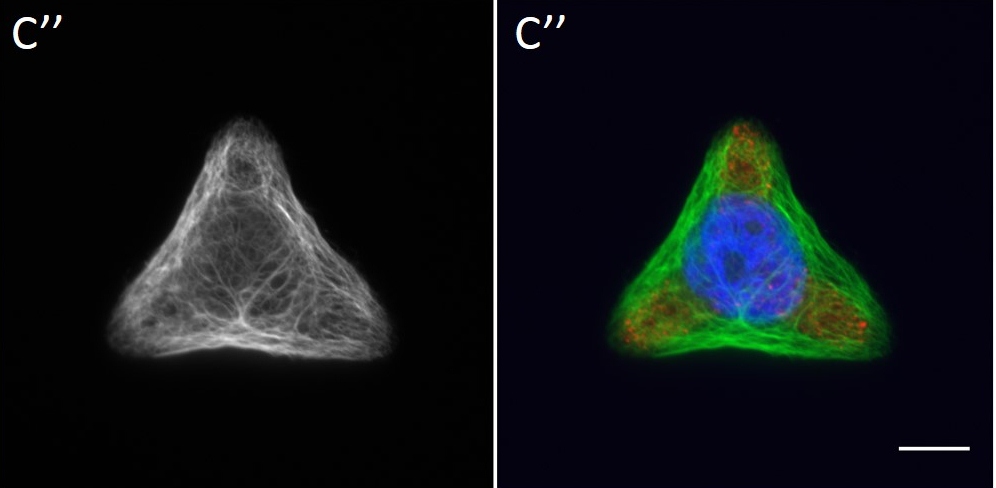
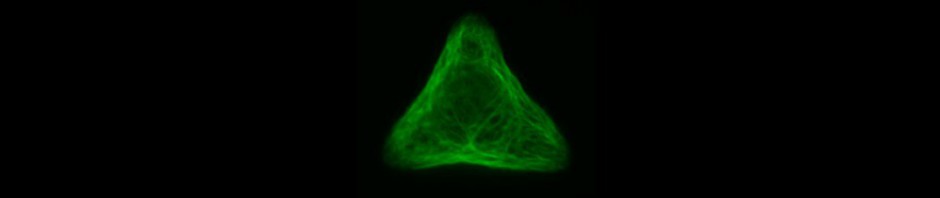
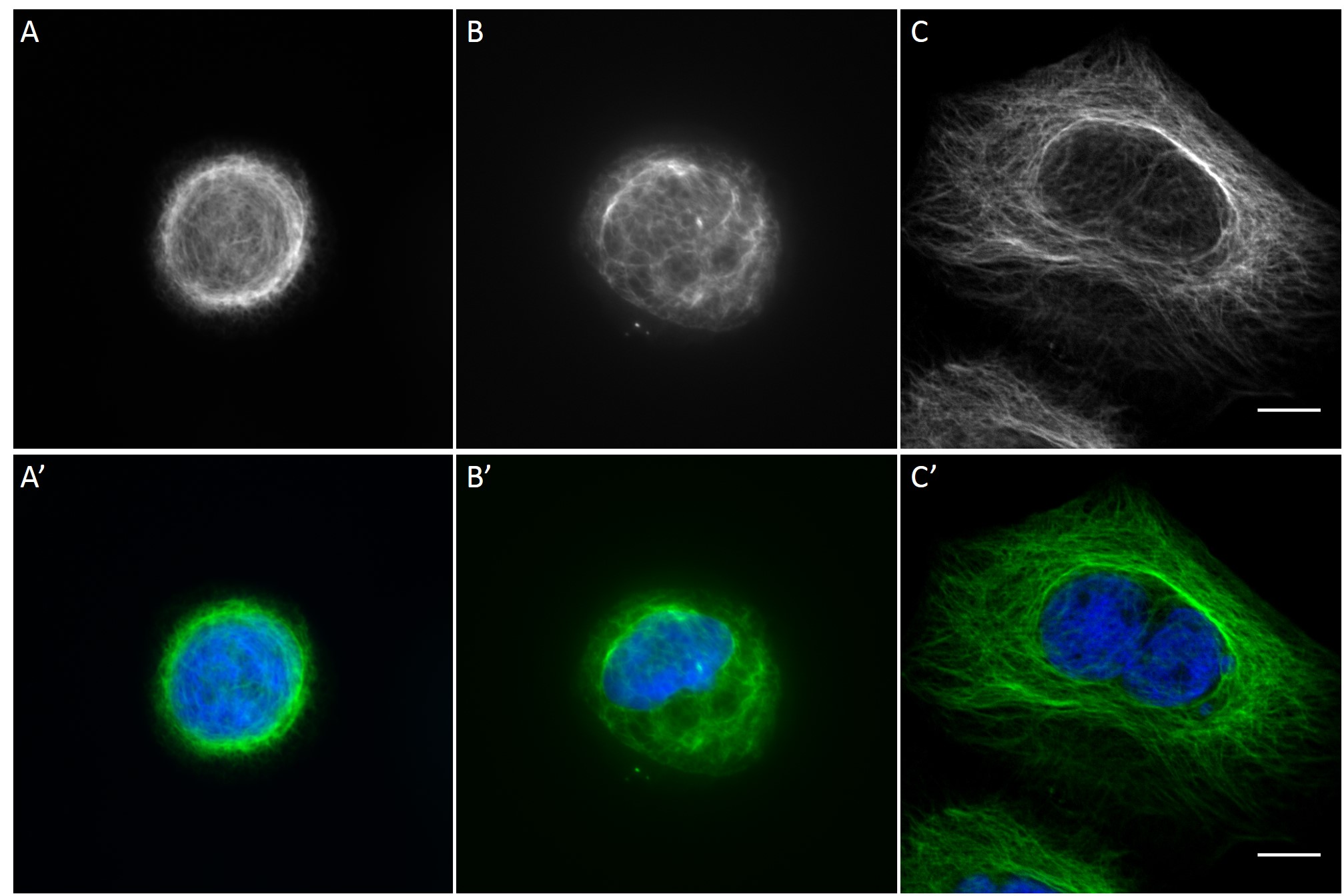
Since in most cases only a small number of normalized cells existed the distribution of human keratin 5 filaments in HaCaT B10 cells and human keratin 13 filaments in AK13-1 cells was assessed based on individual cell images. Independent of the pattern shape and size both keratin filament networks formed a dense network braiding the nucleus and radiated toward the cell periphery. Compared to the compact perinuclear network a decreasing toward the corners was observed, particularly evident in teardrop-shaped (Fig. 13 A”), triangle-shaped (Fig. 13 C”, 15 B’ and 15 C’), square-shaped (Fig. 14 D’) and arrow-shaped (Fig. 14 C’) cells.
![Fig. 13 Individual examples of HaCaT B10 cells spread on 700 µm² micropatterns. HaCaT B10 cells were seeded on a CYTOOchip™ and allowed to spread on the micropatterns for 24 h before fixation. Micropatterns are detected by fluorescently labeled FN (A, B and C), nuclei by DAPI staining (superposed on contrast image [TL] in A', B' and C') and human keratin 5 through EYFP signal (A'', B'' and C''). A merged image of fluorescently labeled FN, DAPI and human keratin 5-EGFP is shown in A''', B''' and C'''. Bar, 10 µm.](http://blog.skythief.de/wp-content/uploads/2015/12/Fig.-13-Individual-examples-of-HaCaT-B10-cells-spread-on-700-µm2-micropatterns.jpg)
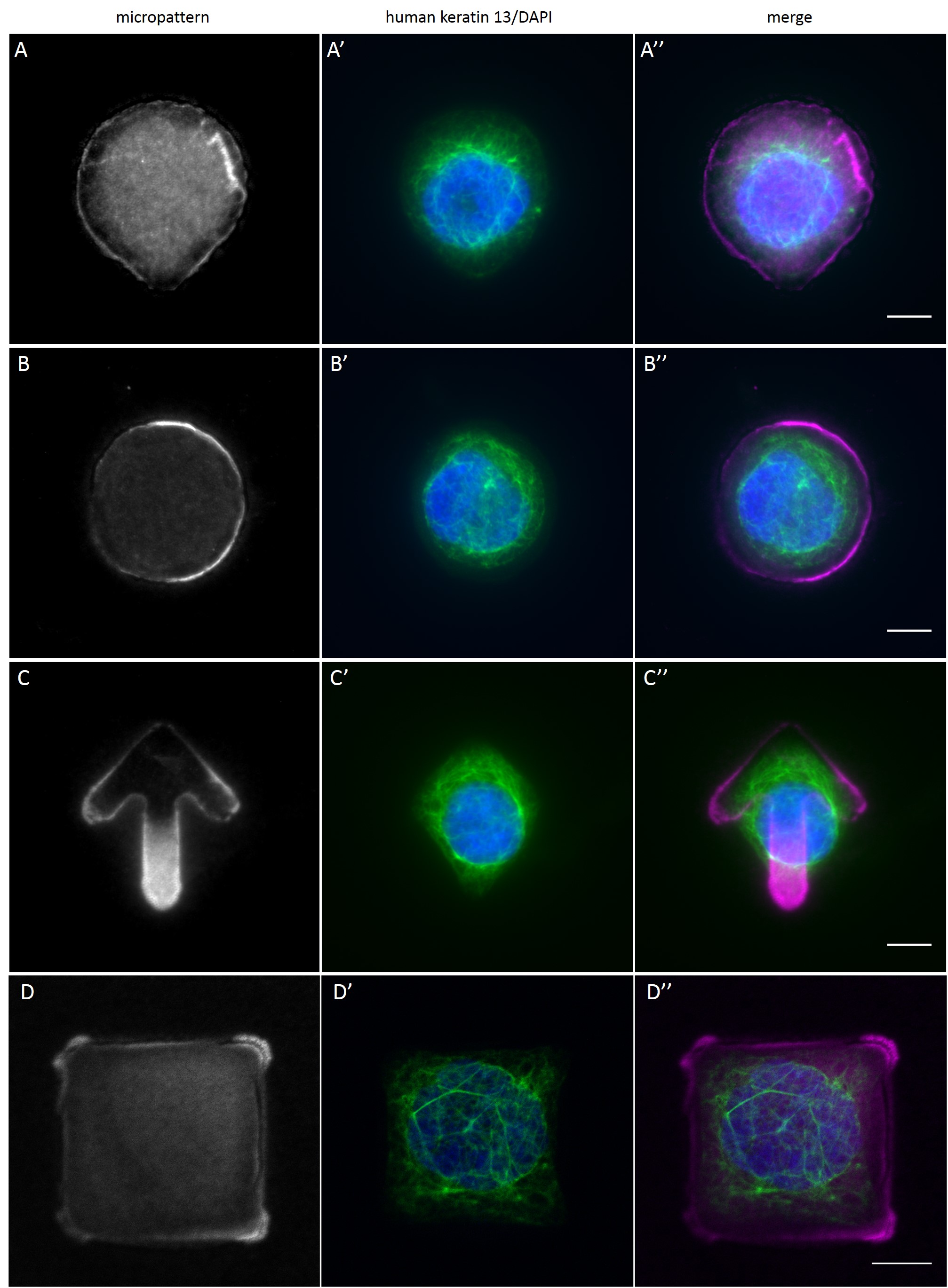
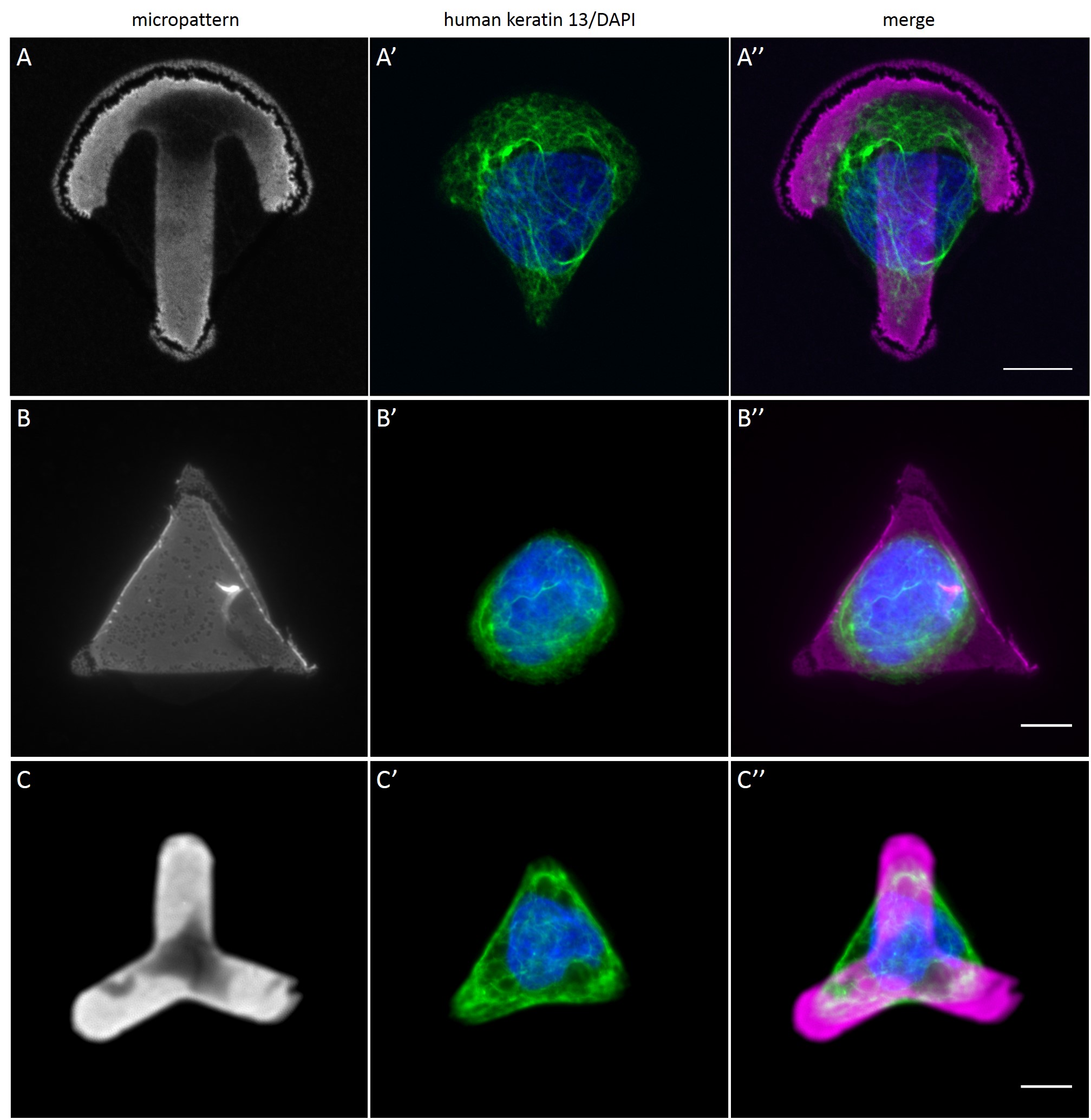
While the average distribution of human keratin 5 in HaCaT B10 cells could quantitatively assessed by generating heat maps of cells spread on crossbow-shaped (Fig. 16 A), disc-shaped (Fig. 16 B) and Y-shaped (Fig. 16C) micropatterns with an average cell spreading area of 700 µm², the average distribution of human keratin 13 in AK13-1 cells was assessed by generating heat maps of cells spread on teardrop-shaped (Fig. 16 E), disc-shaped (Fig. 16 F), arrow-shaped (Fig. 16 G) and square-shaped (Fig. 16 H) micropatterns with an average cell spreading area of 1100 µm².
Images were exported into ImageJ and analyzed with CYTOO tooL for Image Processing-Reference cell, an ImageJ macro provided by CYTOO Inc. Images were aligned, stacked, averaged and pseudo-colored to represent regions of high and low intensity where blue hues represent low frequency of protein localization and red hues represent high frequency of protein localization. If necessary, the macro was adjusted for individual image stacks so that it could identify the corresponding fluorescent micropatterns in order to delineate regions centered on micropatterns and to realign the image stack in all channels.
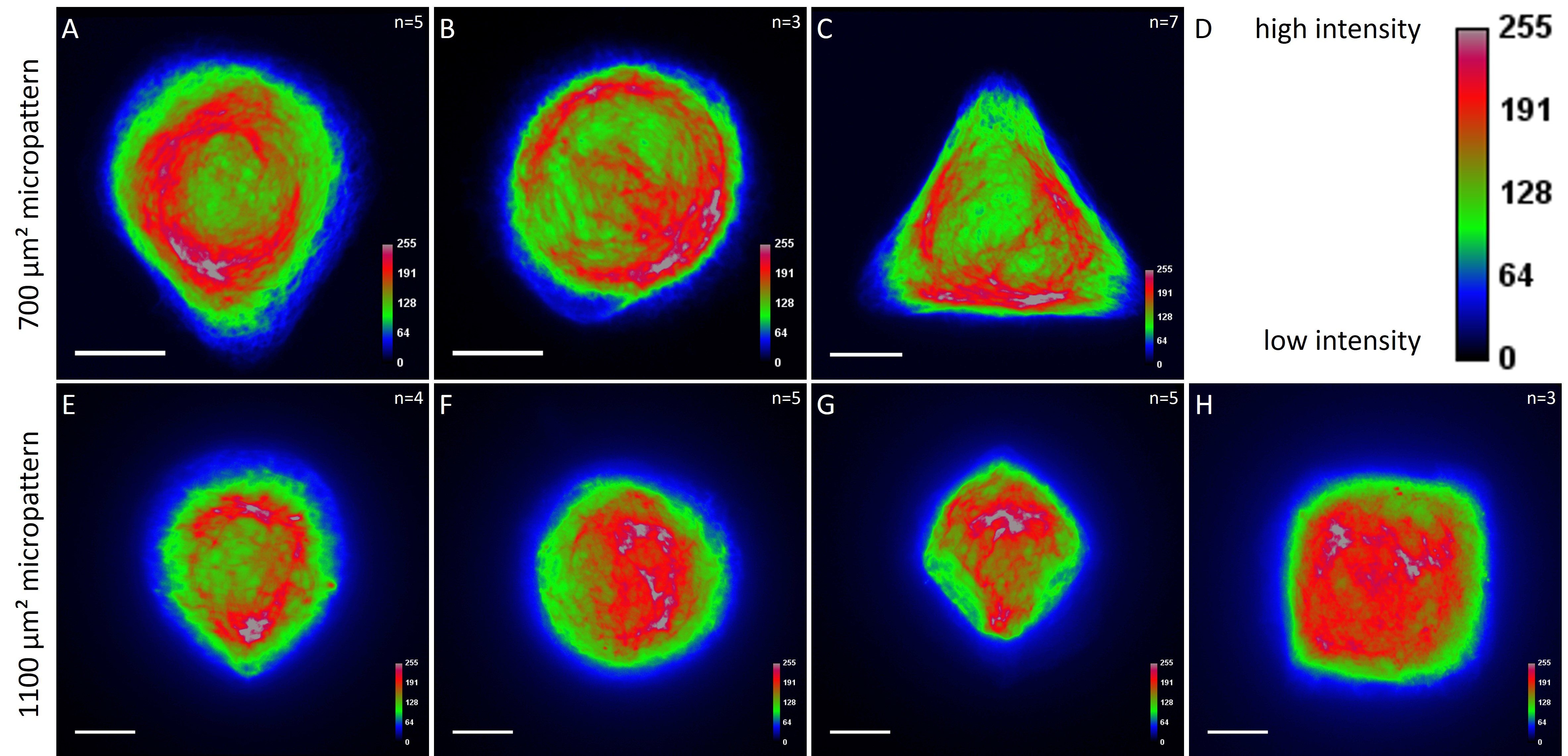
The observation of the average distribution of human keratin 5 in HaCaT B10 cells and of human keratin 13 in AK13-1 cells as seen in individual cell images (Fig. 13-15) could be confirmed for the corresponding shapes. A high average distribution of the KIFs could be observed around the nuclei together with a decrease of the average distribution of the KIFs toward the cell periphery.
[nextpage title=”4 Discussion”]
4 Discussion
Cell micropatterning is becoming a powerful tool for controlling the cellular microenvironment and shape and for studying the effects of physical cues on cellular behavior. Cell micropatterning offers many advantages over growing cells in flasks or in culture dishes according to standard cell culture methods. Micropatterning cells, so as to force them to fit in a certain geometric pattern and size, enables to analyze cellular behavior and to facilitate reliable and reproducible statistical analyses in small scales (~10-50 cells). The objective of this work was to establish a cell micropatterning method for patterning adhesion proteins like FN in predefined geometries on substrates and to analyze the cytoskeleton of single cells grown on these micropatterns.
4.1 Stencil patterning as the protein patterning method of choice
Two methods were testes for patterning FN on substrates: µCP on glass substrates and stencil patterning on silicone elastomer substrates. Stencil patterning showed a significantly better performance than µCP in fabricating accurately shaped micropatterns with a uniform and homogeneous distribution of FN, which constituted the basis for subsequent experiments in the scope of this work and for future experiments.
4.2 Small micropatterns are suitable for single cell analyses
In the next step, the behavior of cells on micropatterned silicone elastomer substrates was analyzed. On average more single cells were found on micropatterns with an average cell spreading area of 700 µm² than on larger micropatterns. This is due to the fact that an attached cell tends to migrate within a large micropattern without fully adapting to its shape and size (Huang et al., 2005; Théry, et al., 2006; Tseng et al., 2012). In the worst case, with time the cell divides, resulting in multiple cells per micropattern. For quantitative analyses of the cytoskeleton of single cells micropatterns with an average cell spreading area of 700 µm² are to be preferred for the cell lines used within this work.
4.3 KIFs show a perinuclear distribution in normalized cells
For quantitatively assessing the distribution of KIFs, heat maps were generated in a number of cases. Since in most cases only a small number of cells of exactly the same size and shape existed the distribution was assessed based of individual cell images. Analyses of the KIF network showed a perinuclear distribution and a decrease toward the corners, irrespective of cell shape and size. Heat maps of selected cell shapes confirmed the observation that the KIF localization was highest around the nucleus and lowest at the edges. These findings indicate that KIFs do not respond to geometrical cues. Similar results were shown for vimentin intermediate filaments (Shabbir et al., 2014). The VIF network of NIH/3T3 cells patterned on differently shaped micropatterns (700 µm²) showed a mostly perinuclear distribution, a decrease toward the cell periphery and an absence in regions marked by sharp edges.
However, a large-scale analysis with more normalized single cells is essential to confirm these first results and to produce a robust statistic, which will provide a more comprehensive view of the biological role of geometrical cues on the behavior of KIFs.
Irrespective of the meaning of geometrical influences on the KIF network, normalized cells enable to build density maps of protein distribution and hence to construct a reference cell, which can serve as a template for identifying cytoskeletal alternations in mutant cells to decipher the role of different molecular components in regulating the cytoskeleton. Future analyses with micropatterned substrates should explore the interplay of KIFs with microfilaments and microtubules in single, dual and triple filament depleted cells.
Even smallest changes in the keratin motion and turnover in response to regulatory cues can now be quantitatively assessed in great detail with less effort than before (Moch et al., 2013).
Furthermore, the use of micropatterns can be an efficient setup to analyze the role of cell-ECM interactions. Take, for example, Y-shaped micropatterns that show ECM-rich regions and regions devoid of ECM, which providing stringent and stationary boundary conditions to cells. Previous studies showed that keratin filament nucleation starts in the cell periphery in close vicinity to focal adhesions (Windoffer et al., 2006), which provide structural links between the ECM and the cytoskeleton. Therefore, it would be interesting to analyze the distribution of focal adhesions in cells spread on Y-shaped micropatterns in order to find out whether the distribution of focal adhesions is restricted to the cell periphery that co-localizes with the edges of the Y-shaped micropattern or if it focal adhesions are also localized in peripheral regions that are in no contact with the ECM of the Y-shaped micropattern. Since focal adhesions connect the ECM to the cytoskeleton, localization is only to be expected in the cell periphery that co-localizes with the ECM-rich regions of the Y-shaped micropattern. Thus, the interplay between focal adhesions, keratin filament nucleation sites and the keratin cycle dynamics can be explored.
4.4 Challenging tasks for future analyses
For analyzing single keratin filaments and keratin bundles, a high resolution and an appropriate spreading of the KIF network have to be achieved. For this purpose, cells have to reach a minimum size in diameter that would allow an appropriate spreading of the cytoskeleton and hence an improved discrimination of single filaments within the meshwork. This means that cells have to be cultured on large micropatterns. This will be a challenging task as the current work demonstrated that an increase in micropattern size led to an increase in the number of cells per micropattern or to incomplete cell spreading of single cells. Epithelial cells tend to clump together since adhesion between cells through cadherins is favored over adhesion to fibronectin through integrins, resulting in multiple cells occupying a single micropattern. In order to enable a high yield of single cells attached and fully spread on large micropatterns, new approaches have to be explored and implemented. The focus is to overcome a few major obstacles. First, special attention has to be given to isolating single cells during the first steps of dispersing the initial cell suspension. Secondly, single cells tend to migrate within large micropatterns without reaching a steady-state position. Furthermore, most single cells on large micropatterns do not spread completely in an appropriate time before cell division. A possible solution to these problems is to design and to test new patterns, which would guarantee the attachment of a single cell and a stable positioning of cells to prevent cell migration. Many previous studies already encountered this problem (Théry et al., 2006; Huang et al., 2005; Tseng et al., 2012). E.g. Tseng et al. (2012) explored the influence of different micropattern configurations on the degree of stability of intercellular junction positioning of cell doublets. Among other findings, the study demonstrated that migration of cell doublets on particular micropattern configurations was impaired. Interestingly, micropattern configurations, which consist of different combinations of adhesive and non-adhesive areas, seem to be promising for controlling cell positioning. Additionally, these micropatterns offer less area for cell attachment thus minimizing the probability of the attachment of multiple cells. Designing micropatterns with different configurations of Y-shaped and X-shaped micropatterns ought to be seriously considered if cells with a substantial diameter are needed.
Although bowtie shaped patterns were designed for the analyses of cell doublets they were not tested as it lay beyond the scope of the present work. Nonetheless, several cell doublets were observed and a few cell doublets were documented on H-shaped micropatterns of the CYTOOchips™ (Fig. 17). It was observed that H-shaped micropatterns provided a highly stable configurations in which each cell was positioned on each side of the gap.
![Fig. 17 Cell pairs on an H-shaped micropattern. 24 h after seeding AK13-1 cells of a CYTOOchip™ cell doublets coupled through desmosomes in the middle are found frequently on H-shaped micropatterns. Micropattern is detected by fluorescently labeled FN (A), nuclei by DAPI staining (superposed on contrast image [TL] in A') and human keratin 13 through EGFP-signal (A''). A merged image of fluorescently labeled FN, DAPI and human keratin 13-EGFP is shown in A'''. Bar, 10 µm](http://blog.skythief.de/wp-content/uploads/2015/12/Fig.-17-Cell-pairs-on-an-H-shaped-micropattern.jpg)
In conclusion, as a result of this work a setup for cell micropatterning has been successfully established. It was determined that the cell lines used in the scope of this work reached a steady-state position on micropatterns with a small cell spreading area and a micropattern configuration consisting of different combinations of adhesive and non-adhesive areas. First results indicate that the KIF network does not respond to local geometrical cues since the distribution of the keratin filaments looks strikingly similar on different micropatterns. A more extensive analysis of the keratin filament distribution is necessary for a robust statistic and for building a reference cell. The results of this work paves the way for analyzing the interplay of the KIF network with other cellular components under controlled microenvironmental parameters.
[nextpage title=”5 Literature”]
5 Literature
-Blanchoin, Laurent, Rajaa Boujemaa-Paterski, Cecile Sykes and Julie Plastino. “Actin Dynamics, Architecture, and Mechanics in Cell Motility.” Physiological Reviews 94, no. 1 (2014): 235-263.
-Chen, C. S., M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber. “Geometric Control of Cell Life and Death.” Science 276, no. 5317 (1997): 1425-1428.
-Coulombe, Pierre A., Michelle L. Kerns and Elaine Fuchs. “Epidermolysis Bullosa Simplex: A Paradigm for Disorders of Tissue Fragility.” Journal of Clinical Investigation 119, no. 7 (2009): 1784-1793.
-de Juan-Pardo, Elena M., Mike Bao-Trong Hoang and Irina M. Conboy. “Geometric Control of Myogenic Cell Fate.” International Journal of Nanomedicine 1, no. 2 (2006): 203-212.
-Fletcher, Daniel A. and Dyche Mullins. “Cell Mechanics and the Cytoskeleton.” Nature 463, no. 7280 (2010): 485-492.
-Fudge, D. S., K. H. Gardner, V. T. Forsyth, C. Riekel and J. M. Gosline. “The Mechanical Properties of Hydrated Intermediate Filaments: Insights from Hagfish Slime Threads.” Biophysical Journal 85, no. 3 (2003): 2015-2027.
-Fudge, Douglas, David Russell, Dan Beriault, Whitney Moore, E. Birgitte Lane and A. Wayne Vogl. “The Intermediate Filament Network in Cultured Human Keratinocytes Is Remarkably Extensible and Resilient.” Plos One 3, no. 6 (2008).
-Hersch, Nils, Benjamin Wolters, Georg Dreissen, Ronald Springer, Norbert KirchgeSsner, Rudolf Merkel and Bernd Hoffmann. “The Constant Beat: Cardiomyocytes Adapt Their Forces by Equal Contraction Upon Environmental Stiffening.” Biology open 2, no. 3 (2013): 351-61.
-Hesse, M., T. M. Magin and K. Weber. “Genes for Intermediate Filament Proteins and the Draft Sequence of the Human Genome: Novel Keratin Genes and a Surprisingly High Number of Pseudogenes Related to Keratin Genes 8 and 18.” Journal of Cell Science 114, no. 14 (2001): 2569-2575.
-Huang, S., C. P. Brangwynne, K. K. Parker and D. E. Ingber. “Symmetry-Breaking in Mammalian Cell Cohort Migration During Tissue Pattern Formation: Role of Random-Walk Persistence.” Cell Motility and the Cytoskeleton 61, no. 4 (2005): 201-213.
-Huang, S. and D. E. Ingber. “Shape-Dependent Control of Cell Growth, Differentiation, and Apoptosis: Switching between Attractors in Cell Regulatory Networks.” Experimental Cell Research 261, no. 1 (2000): 91-103.
-Hyder, Claire L., Kimmo O. Isoniemi, Elin S. Torvaldson and John E. Eriksson. “Insights into Intermediate Filament Regulation from Development to Ageing.” Journal of Cell Science 124, no. 9 (2011): 1363-1372.
-Ingber, D. E., D. Prusty, Z. Q. Sun, H. Betensky and N. Wang. “Cell Shape, Cytoskeletal Mechanics, and Cell Cycle Control in Angiogenesis.” Journal of Biomechanics 28, no. 12 (1995): 1471-1484.
-Janmey, P. A., U. Euteneuer, P. Traub and M. Schliwa. “Viscoelastic Properties of Vimentin Compared with Other Filamentous Biopolymer Networks.” Journal of Cell Biology 113, no. 1 (1991): 155-160.
-Janmey, Paul A. and Christopher A. McCulloch. “Cell Mechanics: Integrating Cell Responses to Mechanical Stimuli.” Annual Review of Biomedical Engineering 9, (2007): 1-34.
-Jonkman, M. F., A. M. G. Pasmooij, Sgma Pasmans, M. P. van den Berg, H. J. ter Horst, A. Timmer and H. H. Pas. “Loss of Desmoplakin Tail Causes Lethal Acantholytic Epidermolysis Bullosa.” American Journal of Human Genetics 77, no. 4 (2005): 653-660.
-Kim, Seyun and Pierre A. Coulombe. “Intermediate Filament Scaffolds Fulfill Mechanical, Organizational, and Signaling Functions in the Cytoplasm.” Genes & Development 21, no. 13 (2007): 1581-1597.
-Kreplak, Laurent and Douglas Fudge. “Biomechanical Properties of Intermediate Filaments: From Tissues to Single Filaments and Back.” Bioessays 29, no. 1 (2007): 26-35.
-Krieg, M., Y. Arboleda-Estudillo, P. H. Puech, J. Kaefer, F. Graner, D. J. Mueller and C. P. Heisenberg. “Tensile Forces Govern Germ-Layer Organization in Zebrafish.” Nature Cell Biology 10, no. 4 (2008): 429-U122.
-Lane, E. B. and W. H. I. McLean. “Keratins and Skin Disorders.” Journal of Pathology 204, no. 4 (2004): 355-366.
-LeDuc, P. R. and R. M. Bellin. “Nanoscale Intracellular Organization and Functional Architecture Mediating Cellular Behavior.” Annals of Biomedical Engineering 34, no. 1 (2006): 102-113.
-Moch, Marcin, Gerlind Herberich, Til Aach, Rudolf E. Leube and Reinhard Windoffer. “Measuring the Regulation of Keratin Filament Network Dynamics.” Proceedings of the National Academy of Sciences of the United States of America 110, no. 26 (2013): 10664-10669.
-Pallari, Hanna-Mari and John E. Eriksson. “Intermediate Filaments as Signaling Platforms.” Science’s STKE : signal transduction knowledge environment 2006, no. 366 (2006): pe53-pe53.
-Parker, K. K., A. L. Brock, C. Brangwynne, R. J. Mannix, N. Wang, E. Ostuni, N. A. Geisse, J. C. Adams, G. M. Whitesides and D. E. Ingber. “Directional Control of Lamellipodia Extension by Constraining Cell Shape and Orienting Cell Tractional Forces.” Faseb Journal 16, no. 10 (2002).
-Provenzano, Paolo P. and Patricia J. Keely. “Mechanical Signaling through the Cytoskeleton Regulates Cell Proliferation by Coordinated Focal Adhesion and Rho Gtpase Signaling.” Journal of Cell Science 124, no. 8 (2011): 1195-1205.
-Ramms, Lena, Gloria Fabris, Reinhard Windoffer, Nicole Schwarz, Ronald Springer, Chen Zhou, Jaroslav Lazar, Simone Stiefel, -Nils Hersch, Uwe Schnakenberg, Thomas M. Magin, Rudolf E. Leube, Rudolf Merkel and Bernd Hoffmann. “Keratins as the Main Component for the Mechanical Integrity of Keratinocytes.” Proceedings of the National Academy of Sciences of the United States of America 110, no. 46 (2013): 18513-18518.
-Rugg, E. L. and I. M. Leigh. “The Keratins and Their Disorders.” American Journal of Medical Genetics Part C-Seminars in Medical Genetics 131C, no. 1 (2004): 4-11.
-Russell, D., P. D. Andrews, J. James and E. B. Lane. “Mechanical Stress Induces Profound Remodelling of Keraitin Filaments and Cell Junctions in Epidermolysis Bullosa Simplex Keratinocytes.” Journal of Cell Science 117, no. 22 (2004): 5233-5243.
-Shabbir, Shagufta H., Megan M. Cleland, Robert D. Goldman and Milan Mrksich. “Geometric Control of Vimentin Intermediate Filaments.” Biomaterials 35, no. 5 (2014): 1359-1366.
-Shimizu, A., A. Ishiko, T. Ota, K. Tsunoda, M. Amagai and T. Nishikawa. “Igg Binds to Desmoglein 3 in Desmosomes and Causes a Desmosomal Split without Keratin Retraction in a Pemphigus Mouse Model.” Journal of Investigative Dermatology 122, no. 5 (2004): 1145-1153.
-Stiles, Jessica M., Robert Pham, Rebecca K. Rowntree, Clarissa Amaya, James Battiste, Laura E. Boucheron, Dianne C. Mitchell and Brad A. Bryan. “Morphological Restriction of Human Coronary Artery Endothelial Cells Substantially Impacts Global Gene Expression Patterns.” Febs Journal 280, no. 18 (2013): 4474-4494.
-Strnad, P., R. Windoffer and R. E. Leube. “Induction of Rapid and Reversible Cytokeratin Filament Network Remodeling by Inhibition of Tyrosine Phosphatases.” Journal of Cell Science 115, no. 21 (2002): 4133-4148.
-Thery, M., A. Pepin, E. Dressaire, Y. Chen and M. Bornens. “Cell Distribution of Stress Fibres in Response to the Geometry of the Adhesive Environment.” Cell Motility and the Cytoskeleton 63, no. 6 (2006): 341-355.
-Thery, M., V. Racine, A. Pepin, M. Piel, Y. Chen, J. B. Sibarita and M. Bornens. “The Extracellular Matrix Guides the Orientation of the Cell Division Axis.” Nature Cell Biology 7, no. 10 (2005): 947-U29.
-Tseng, Qingzong, Eve Duchemin-Pelletier, Alexandre Deshiere, Martial Balland, Herve Guillou, Odile Filhol and Manuel Thery. “Spatial Organization of the Extracellular Matrix Regulates Cell-Cell Junction Positioning.” Proceedings of the National Academy of Sciences of the United States of America 109, no. 5 (2012): 1506-1511.
-Windoffer, R., A. Kolsch, S. Woll and R. E. Leube. “Focal Adhesions Are Hotspots for Keratin Filament Precursor Formation.” Journal of Cell Biology 173, no. 3 (2006): 341-348.
-Windoffer, R. and R. E. Leube. “Detection of Cytokeratin Dynamics by Time-Lapse Fluorescence Microscopy in Living Cells.” Journal of Cell Science 112, no. 24 (1999): 4521-4534.
-Windoffer, Reinhard, Michael Beil, Thomas M. Magin and Rudolf E. Leube. “Cytoskeleton in Motion: The Dynamics of Keratin Intermediate Filaments in Epithelia.” Journal of Cell Biology 194, no. 5 (2011): 669-678.